Congenital Adrenal Hyperplasia (CAH)
- autosomal recessive traits.
- The most common enzyme defect is 21-hydroxylase deficiency.
- 11β-hydroxylase & 17-hydroxylasedeficiency → hypertension due to excess production of 11-deoxycorticosterone having mineralocorticoid activity
Clinical features:
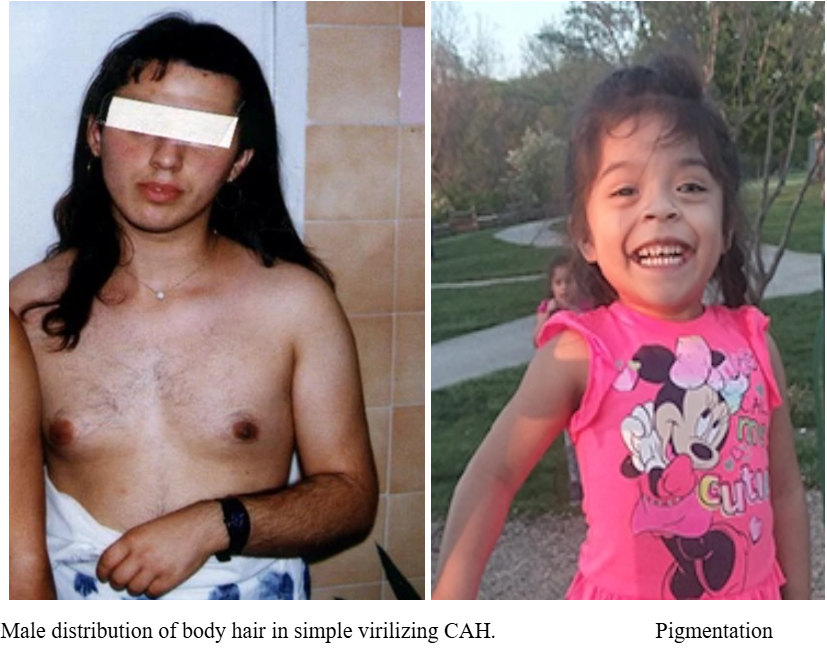
- Ambiguous genitalia in girls (androgen excess)
- Amenorrhoea, hirsutism, pigmentation (ACTH),
- Hypotension, hypertension
- Androgen excess→ precocious pseudopuberty, which is distinguished from ‘true’ precocious puberty by low gonadotrophins.
Investigation
| Investigations | Finding |
|
|
|
|
|
In siblings of affected children
|
|
Management:
- Corticosteroid replacement
- reverse rhythm to suppress early morning ACTH (steroid given at night)
- Late-onset 21-hydroxylase deficiency:
- corticosteroid may not be needed.
- For hirsutism: anti-androgen therapy
Follow up:
- Clinical:
- menstrual cycle, hirsutism,
- weight gain, blood pressure
- biochemical profiles
- plasma renin,
- 17-OH-progesterone,
- testosterone levels